Studying secondary metabolites
While primary metabolites (e.g. lipids, carbohydrates, proteins, nucleic acids) are part of fundamental metabolic pathways essential for survival, secondary metabolites (e.g. alkaloids, antibiotics, phenazines, terpenoids) can provide evolutionarily advantageous features important for an organism’s survival. Our current knowledge of secondary metabolites are only the tip of the iceberg of what biology is capable of producing. As more and more organisms are sequenced, more and more new, unique biosynthetic clusters are revealed each day. The secondary metabolites associated with each have yet to be discovered and functionally characterized.
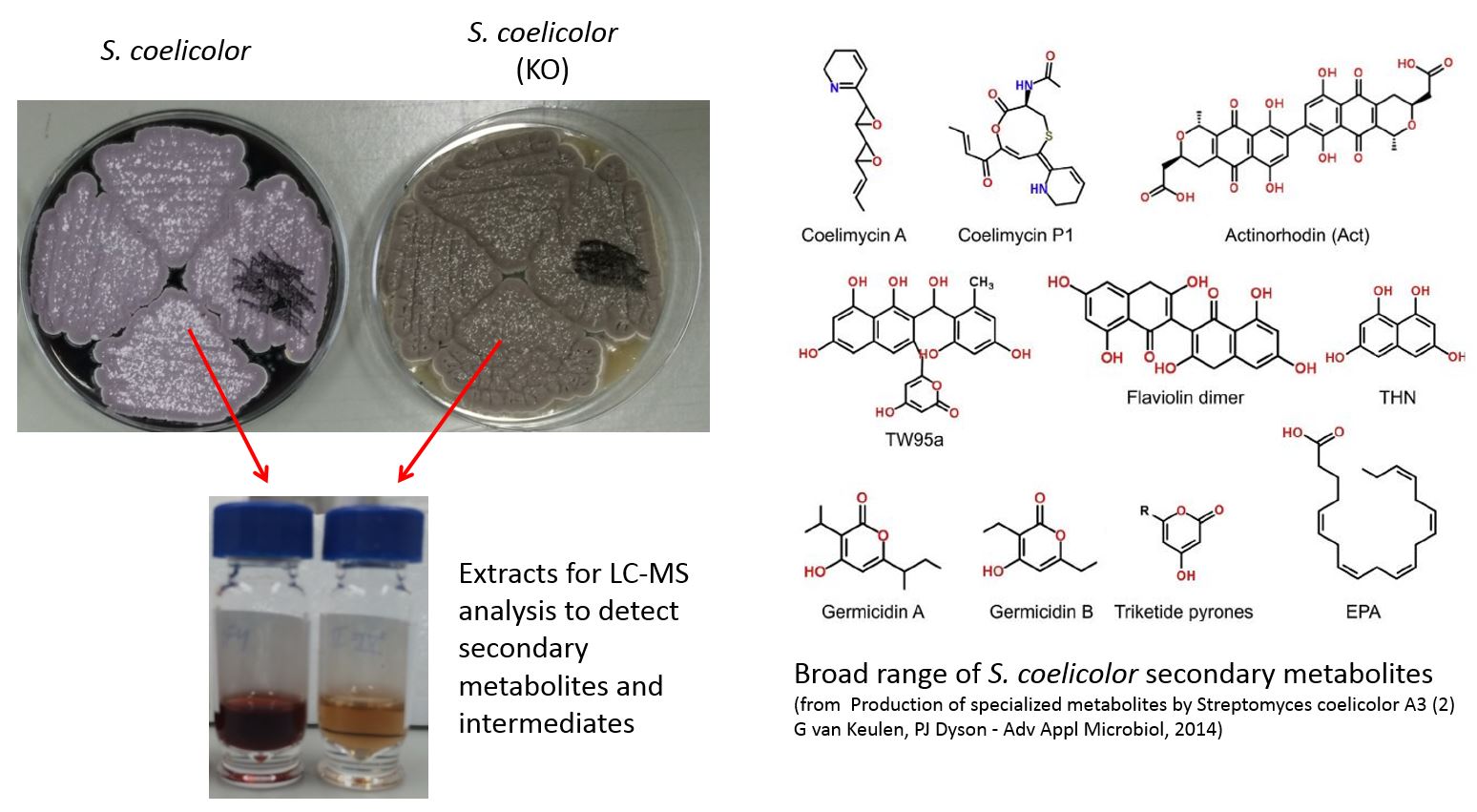
Here, using advanced mass spectrometry-based techniques, secondary metabolites that are produced or consumed by various organisms (plant, fungus, microbe, etc) under various conditions are measured. Characterizing the metabolite profile of organisms provides the opportunity to gain insight and create linkages between sequence and function.
In synthetic biology, this metabolic information is used to determine the relative synthesis of targeted secondary metabolites from different constructs, discover new compounds as they relate to genomic information, as well as identify relative levels of pathway intermediates, shunt products and metabolic precursors to better understand pathway dynamics for improved design.
MIDAS – Overcoming barriers to discover novel secondary metabolites
Major challenges to overcome in secondary metabolite analysis of mass spectrometry data include identification of the metabolite and determining whether the metabolite has already been discovered, or is novel and possibly representing a new class. When a metabolite standard is available to run LC-MS/MS and compare sample mass spectra to standard mass spectra, the answer to this question is simple. However, often standards are not available, and online databases with MS/MS fragmentation data is scarce for secondary metabolites.
MIDAS (Metabolite Identification via Database Searching) is one approach developed by researchers here to begin addressing these challenges1. This is essentially an open-source software tool that predicts MS/MS fragments from metabolites in a database. A metabolite is then identified, with a percent probability, by comparing predicted fragments to actual measured fragments from a sample. In the absence of MS/MS fragmentation data from a standard, this is a powerful tool for metabolite identification and dereplication for discovery of new metabolites.